
In LLPSDB v2.0, a protein with specific sequence alone or with other components (proteins or nucleic acids) validated to undergo LLPS (or NOT) in vitro is curated carefully. The user-guide of “Browse” and “Search” modules is as the following:(Note: Symbol “-” means none, and “N/A” means data are unknown or unsure.)
The curated data are grouped into “Unambiguous system” and “Ambiguous system”.
For “Unambiguous system”, same as LLPSDB v1.0, three classifications can be browsed based on: “protein type”, “main components type” and “main components number”.
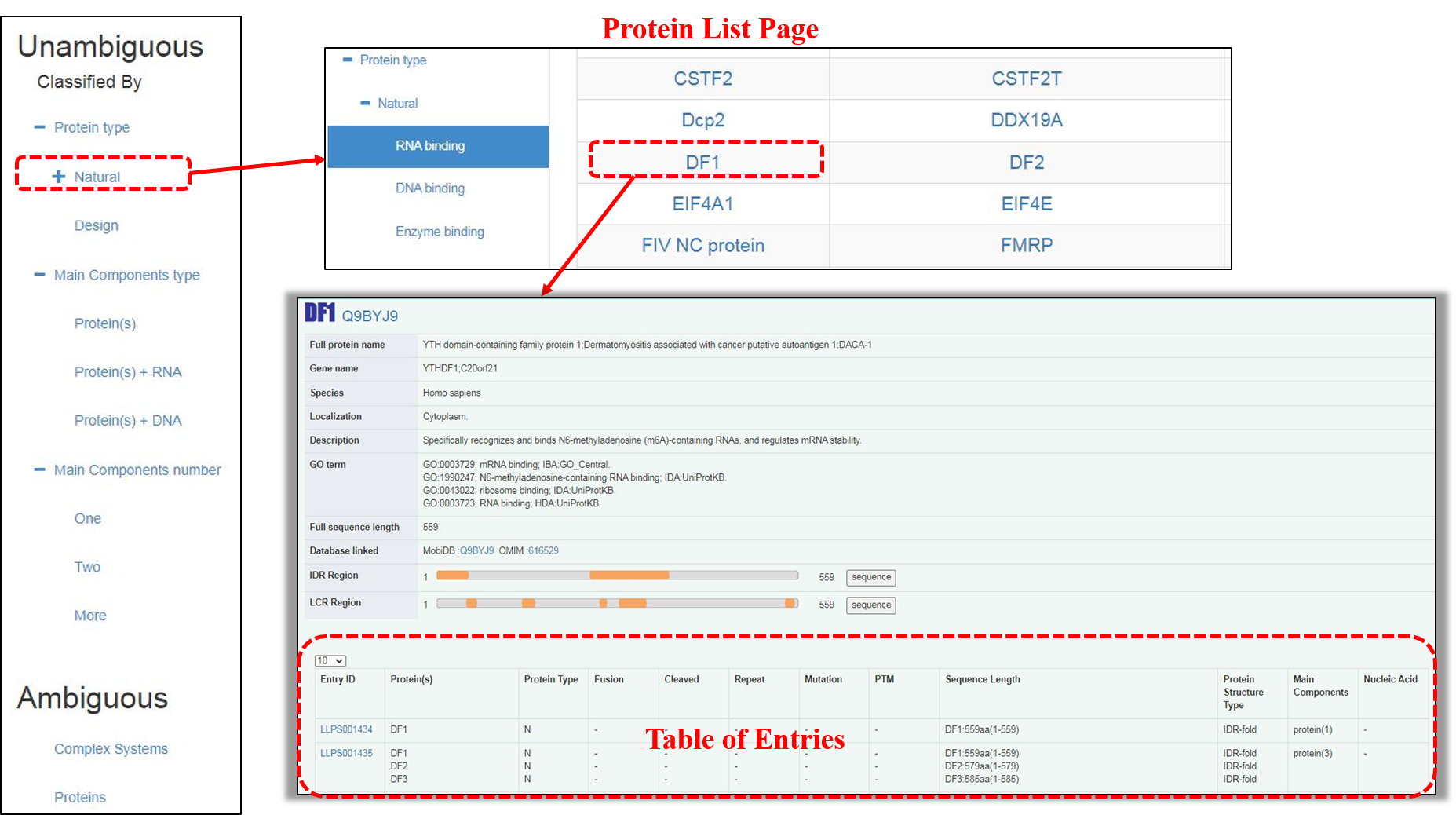
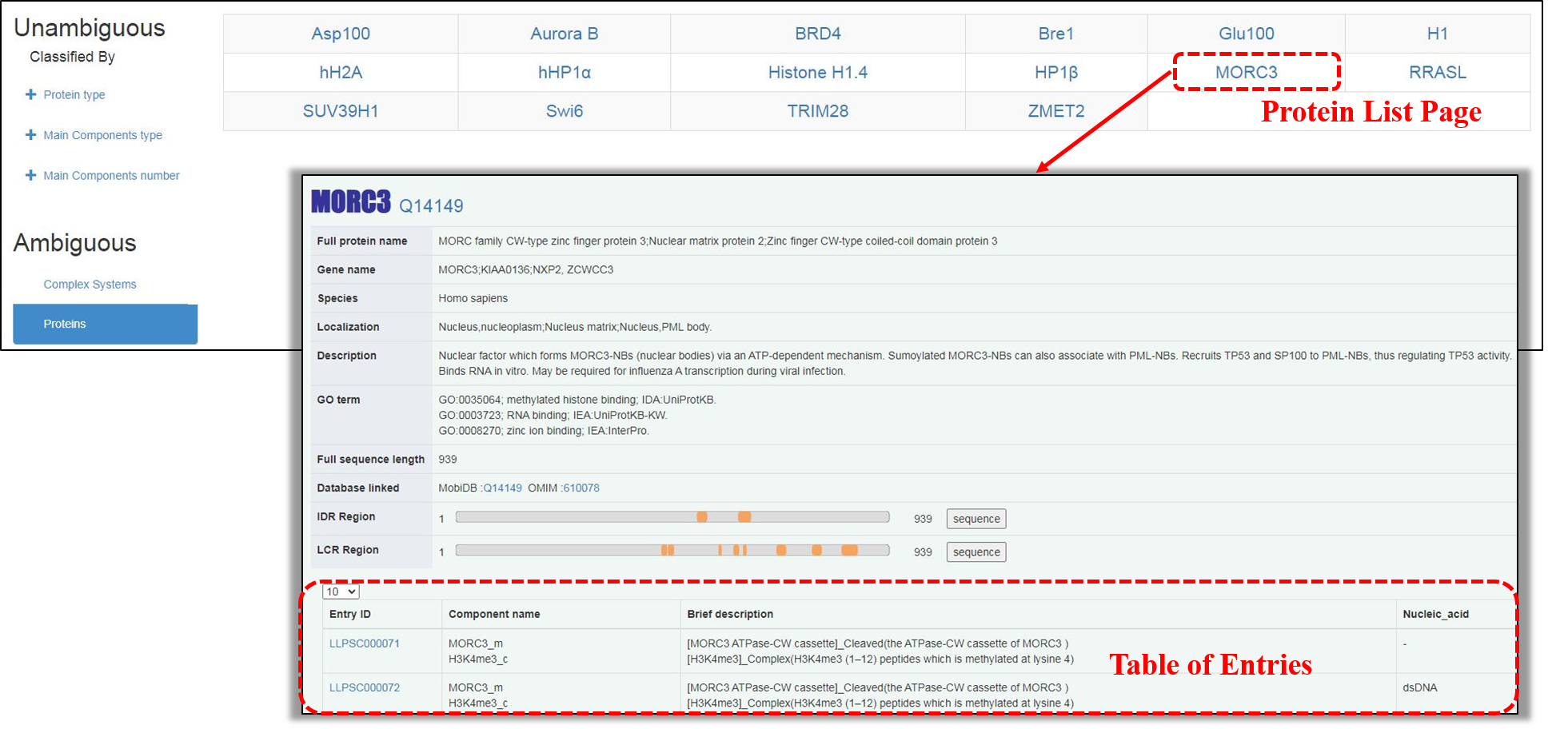
The “protein type” includes two subclasses: independent “Natural” proteins and “Designed” ones. By browsing either of them, an extra Protein List page is displayed at first, which can be only found in this classification. Natural proteins are grouped according to their molecular function annotations in GO term on the page. Upon clicking on the abbreviation of independent protein name, the Protein Details page is accessible, in which the general information of this protein is shown, followed by a “Table of Entries” related to it.
oUniProt ID / NCBI code
oFull protein name
oGene name
oSpecies
oLocalization
oDescription
oGene Ontology (GO) term
oFull sequence length
oDatabase linkage (MobiDB, DisProt, OMIM, IDEAL, FuzDB, AmyProt)
oIDR (intrinsically disordered region) visualization
oLCR (low complexity region) visualization
The IDR and LCR visualizations are only provided for natural proteins. In addition, full protein sequence and the IDRs and the LCRs within them presented in highlight could be found by clicking on the “sequence” buttons following the visualized horizontal brands. IDRs are identified via searching in MobiDB and not shorter than 15 residues. For those sequences not available in MobiDB, the PONDR VL3-BA algorithm is used for prediction (Obradovic Z. et al. 2003, Proteins. 53 Suppl 6, 566-572). LCR is predicted by using the SEG algorithm (Piovesan, D. et al. 1994, Computers & chemistry.18,269-285) with default parameters. The designed proteins are automatically categorized as IDRs.
The “main components type” classifies the data into three subclasses based on whether the condensate includes proteins only, proteins and RNA, or proteins and DNA. The “main components number” classifies the data into systems including one, two, or more components. In these two classifications, “main components” refer to the involved proteins, DNA or RNA. For the experiments that a group of different DNA or RNA were added as a mixture, “RNA” or “DNA” is deposited as a single component, and its internal sequence variance is not considered. Other molecules such as salt, buffer solvent as well as crowding agent are not considered as main components, but “experimental conditions” instead.
“Table of Entries” is provided for each classification. It displays entry ID, protein(s) name, protein type, Fusion, Cleaved, Repeat, Mutation, post-translational modification (PTM), sequence length, protein structure type, main components and nucleic acid(s) of each entry.
Entry ID: An identifier for each entry in a format of “LLPSxxxxxx” where the “xxxxxx” denotes serial number.
Protein(s): Specific protein name. Several symbols are adopted.
| - | A fusion construction |
| _m | Protein with modification(s) (including be cleaved, repeat, mutation and PTM) |
It should be noted that the symbol “-” inherently exists in some specific protein names where in most cases a number follows it. In this database, these protein names are listed below:
| LAF-1 | PSD-95 | TDP-43 | GAR-1 | N-WASP |
| PGL-3 | SLP-76 | Intersectin-1 | SPD-5 | PLK-1 |
| SPD-2 | TPXL-1 | MEX-5 | engrailed-2 | PGL-1 |
| SEPA-1 | EPG-2 | β-amyloid | rmfp-3b | UNC-10 |
| rmfp-1 | HBP-1 | HBP-pep | alpha-tubulin | beta-tubulin |
| Mfp-3S | SYD-2 | ELKS-1 | HBP-2 | fibrillin-1 |
| Mfp3S-pep | Mfp3F-pep | aquaporin-Z | U1-70K | YAP-1β |
| γ-gliadin | Centrin-2 | Alpha-synuclein | TIAR-2 | ELP1-RBD |
| LL-III | N-protein | LL-III | YAP-2α | β-synuclein |
| complexin-1 | β-Pix_mouse | β-Pix_rat | NELF-E | GSK-3 |
| AF-9 | Galectin-3 | LAF-1 RGG shuf | LL-III | GRN-3 |
| GRN-5 | Importin-β | Importin-α | LAF-1 RGG shuf-pres | HTLV-1 NC protein |
| hγ-D-crystallin | rγ-D-crystallin | rmfp-3b-NT | fluorescein-labeled control peptide |
Note: An entry containing multiple rows means a system with multiple proteins, in which each row presents one protein component (for the columns from “Protein(s)” to “Protein structure type”). For a fusion protein, if the corresponding records are not same for each fused part, there will be more than one record in columns “Protein type”/“Cleaved”/“Repeat” separated one by one with symbol “,”.
Protein type: Natural protein(s) are noted as “N” and designed ones as “D”.
Fusion: The fusion protein construct is marked with “Yes”, otherwise with “-”.
Cleaved: If protein is cleaved, it is noted as “Yes”, otherwise as “-”. If the domain removed from or retained in the cleaved protein is known, symbol “( )” is presented instead of “Yes”, within which the domain name is presented, and symbol “△” means the following domain it is removed.
Repeat: Records of the number of repeated region or protein. Symbol “-” means no repeat, and “N/A” means the number of repeat is unknown.
Mutation: “Mn” is an identifier for different mutations of each protein, where the “n” is a serial number for distinguishing mutated site and type. Symbol “-” denotes no mutation.
PTM: Post-translational modification of protein. Phosphorylation, methylation and acetylation are noted as Phos, Meth and Ac respectively. Same as Mutation, the subsequent serial number denotes an identifier for distinguishing different sites that have same type of PTM. Symbol “-” denotes no PTM.
Sequence length: The length and residue region of the protein sequence. If the sequence region of the protein is known, it is recorded within “()” in a form of “n1-n2” in which “n1” means the initial and “n2” means the ending residue number. For fusion proteins, the region or length of each fused part (if it is known) is separated by “;” within “()”, while the linker part is not noted. For example, for entry LLPS001051, “VRN1-PSA-VRN1_m:267aa(1-105;41;221-341)” means the fusion protein has three parts, VRN1, PSA and VRN1, totally 267 amino acids, with the first part VRN1 containing residue 1-105, the second part PSA containing 41 amino acids, and the last part VRN1containing residue 221-341. For dimeric proteins, different subunits are separated by symbol “/”.
Protein structure type: Structure annotation of protein(s). All designed proteins are considered as disordered except two (SpyC1, SpyC2) noted as “fold” in the literature. Structure types of natural proteins recorded in UniProt are identified according to the MobiDB records; for those not recorded in UniProt, their structure types are identified by PONDR VL3-BA (Obradovic et al., 2003). Protein structure types could be “IDR”, “fold” and “IDR-fold”:
IDR: This protein or peptide is intrinsically disordered.
fold: This protein or peptide is folded.
IDR-fold: IDR domain coexists with folded domain in this protein or peptide.
Main components: The type of main components in each entry, which is recorded in the form of “Protein(n) + RNA” or “Protein(n) + DNA” or “Protein(n)”, where the “n” indicates the number of proteins.
Nucleic acid(s): Records of nucleic acid(s) coalesced with protein(s) in system.
“Entry” in LLPSDB v2.0 is identified by main components, which means if two systems contain the same composition of protein(s) and nucleic acid(s), they belong to the same entry. By clicking on the Entry ID in “Table of Entries”, users can access into Entry page. All data on this page are presented in two parts: general information and phase separation conditions.
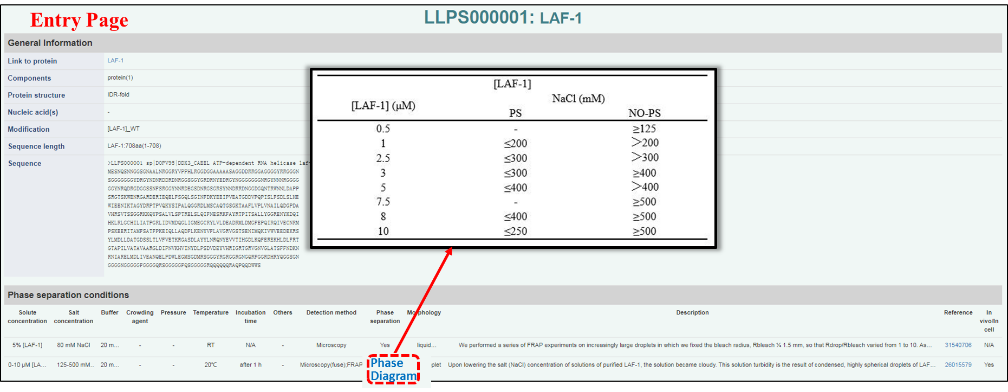
General information :
More specific information for each entry is displayed top part here. Protein details page can be linked from the first line. To be clear, in the description of “Modification” and “Sequence length”, the protein name and/or domain name (or region) are recorded within “[ ]”, and the number adjacent to it is the repeat number of this domain (or region) in the protein.
Modification: Short description about the modification type of the protein(s). For a modified protein, its name and modification type are separated by “_”. More details of the modification are described within “( )”. Several special abbreviations are used here:
| WT | Wild Type |
| Phos | Phosphorylation |
| dePhos | dePhosphorylation |
| Meth | Methylation |
| diMeth | diMethylation |
| hypoMeth | hypoMethylation |
| Ac | Acetylation |
Sequence: Protein sequence(s) is/are displayed in FASTA format on this page (Note that post-translational modifications are not manifested in sequence).
Phase separation conditions:
(Note: The original data and unit retrieved from literatures are deposited.)
Solute concentration: Protein(s) (and nucleic acid(s)) concentration.
Salt concentration: Salt concentration.
Buffer: Buffer solution concentration and pH it maintains.
Crowding agent: Type and concentration of macromolecular crowding agent used to mimic highly crowded cytoplasm environment.
Pressure: Experimental pressure.
Temperature: Experimental temperature (“RT” means room temperature).
Incubation time: Experimental incubation time.
Others: Other specific materials and/or conditions applied in experiments
Detection mothed: The experiment technique used to detect phase separation and characterize the phase morphology. General dynamic liquid droplet indications (flow, fuse, wetting, dropping, reversible) are noted within “()” following the detection method. Several abbreviations are used:
| SEM | Scanning Electron Microscopy |
| TEM | Transmission Electron Microscopy |
| Cryo-EM | Cryo-Electron Microscopy |
| FRAP | Fluorescence Recovery after Photobleaching |
| DLS | Dynamic Light Scattering |
| SDS-PAGE | Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis |
Phase separation: Undergoing LLPS or not. Those noted with “Phase diagram” can be linked to a phase diagram in digital format.
Morphology: A brief annotation of phase morphology. Most are identified as “liquid droplets” or “droplets”, while some gel, fiber or aggregate ripen from liquid droplet are also recorded here with “droplet to gel” or “droplet to fiber” term respectively.
Description: Description of phase separation tendency extracted from literature.
Reference: PubMed ID (or DOI number if PubMed ID is not available) of the literature.
In vivo/In cell: Whether there is related in vivo/in cell data in the literature.
For “Ambiguous system”, the dataset can be browsed through “Complex Systems”. By clicking “Complex Systems”, a brief table (similar to the “Table of Entries” in “Unambiguous system”) is presented and a detailed Entry page can be accessed by clicking Entry ID.
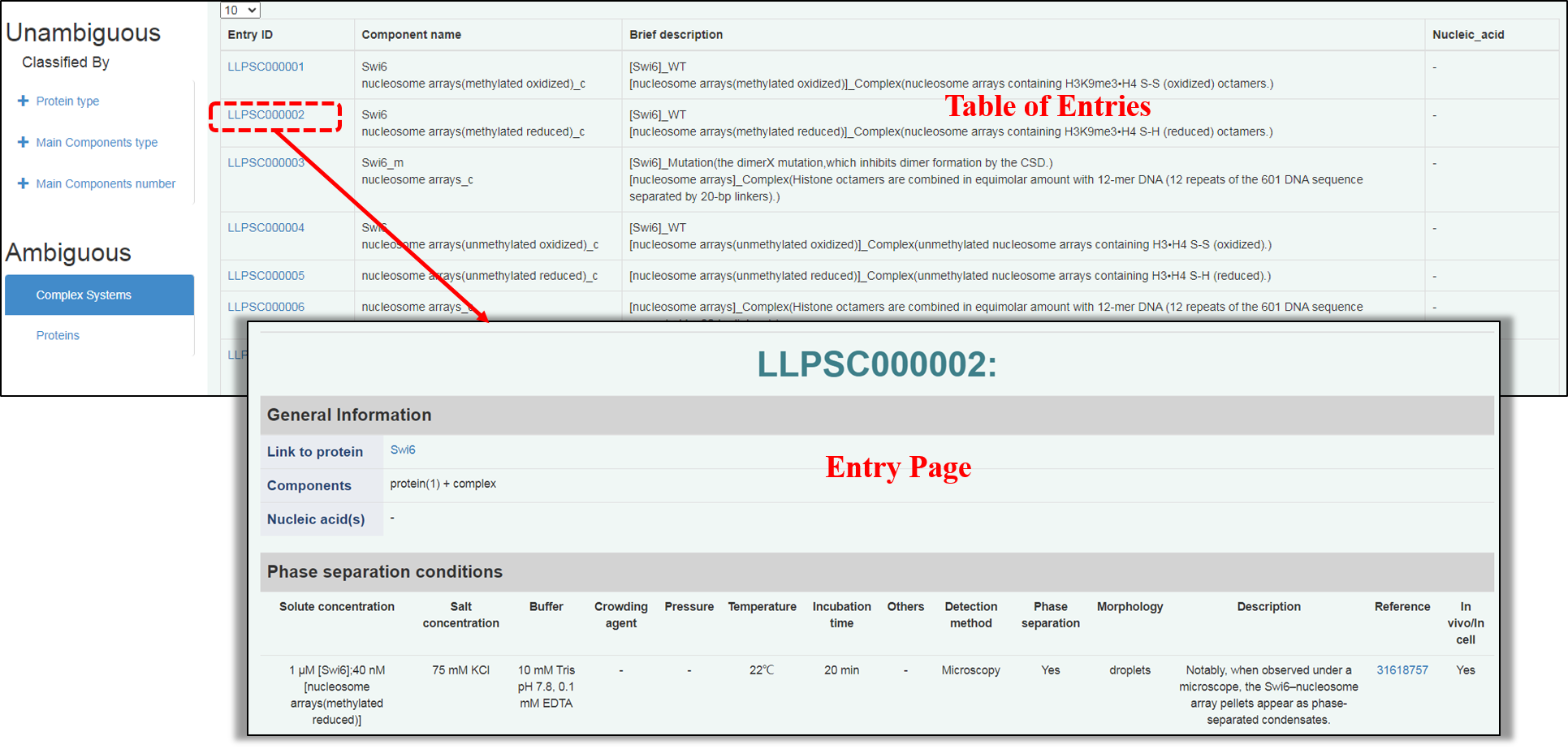
Entry ID: An identifier for each entry in a format of “LLPSCxxxxxx” where the “xxxxxx” denotes serial number.
Component name: The name of components in each entry, including complex mixture with "_c" as a lable(and unambiguous protein in some entries).
Brief description: A brief description of components in a form same as that in "modification" of Entry page in "Unambiguous system".
Nucleic acid(s): Records of nucleic acid(s) coalesced in system.
In Entry page, the experimental conditions are presented as those in “Unambiguous system”.
The proteins with clear information involved in LLPS in“Ambiguous system”can be browsed through the left button“proteins”. Similar with those in “Unambiguous system”, an extra Protein List page is displayed at first. Upon clicking on the abbreviation of independent protein name, the Protein Details page is accessible, in which the general information of this protein is shown, followed by a “Table of Entries”related to it.
For “Search by options”, the searched results are shown in a form as “Table of Entries” which is detailed above in “Browse” module, except that when user select the search option “Phase separation status”, which is a new option in LLPSDB v2.0 VS LLPSDB v1.0. It should be noted that the searching results through this option show the detailed experimental condition as described in “Phase separation conditions” part in “Browse” module.
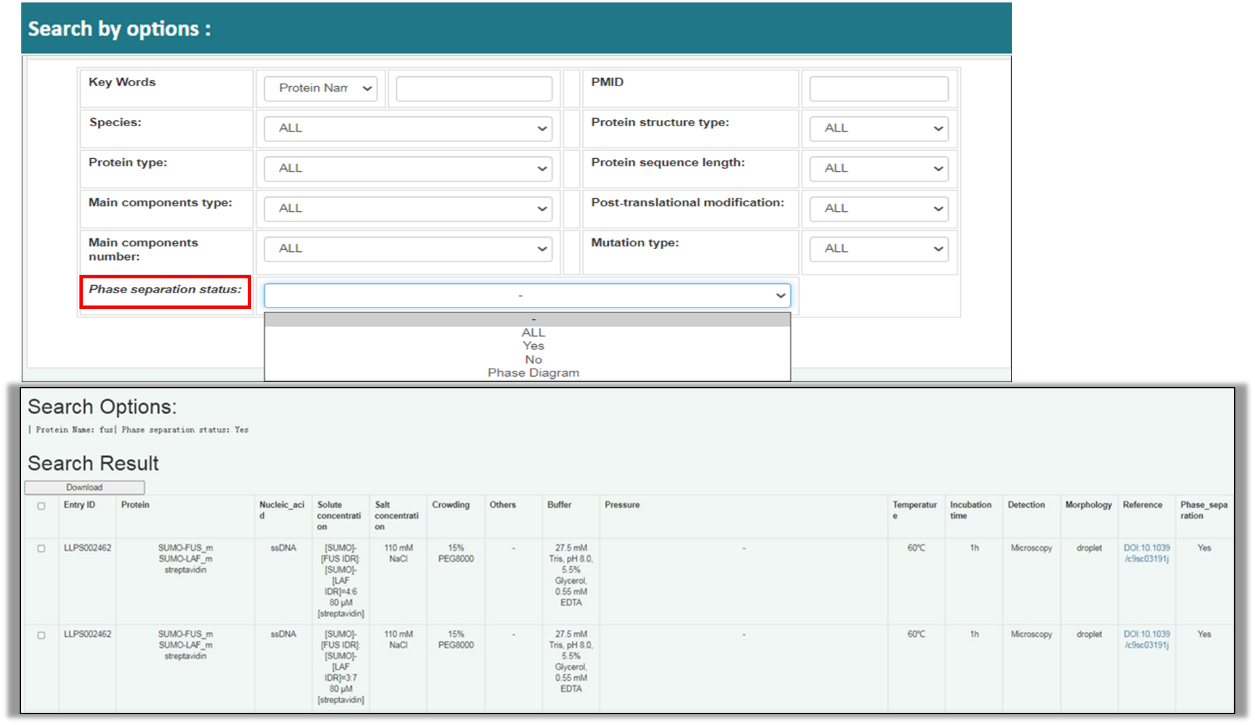
For “Search by protein sequence”, by using “blast”, user can search protein sequence in LLPSDB v2.0 with the default e-value set as 1e-6. The searched results are displayed in ascending order of e-value.
Selected searching results can be downloaded by ticking the check boxes on the most left side. All searching results can be downloaded by clicking on“download” on the topside.The downloaded CSV files are encoded in Unicode (UTF-8).
By clicking on the searched Entry ID, user also can access into the Entry page.
It should be noted that "Ambiguous system" can not be searched in LLPSDB v2.0.
Piovesan, D., Necci. M., Escobedo, N., Monzon, AM., Hatos, A., Mičetić, I., Quaglia, F., Paladin, L., Ramasamy, P., Dosztányi, Z., Vranken, WF., Davey, NE., Parisi, G., Fuxreiter, M., Tosatto, SCE. (2021). MobiDB: intrinsically disordered proteins in 2021. Nucleic Acids Res 49(D1), D361-D367.
Obradovic, Z., Peng, K., Vucetic, S., Radivojac, P., Brown, C.J., and Dunker, A.K. (2003). Predicting intrinsic disorder from amino acid sequence. Proteins 53 Suppl 6, 566-572.
Piovesan, D., Tabaro, F., Paladin, L., Necci, M., Micetic, I., Camilloni, C., Davey, N., Dosztanyi, Z., Wootton, J.C. (1994). Non-globular domains in protein sequences: automated segmentation using complexity measures. Computers & chemistry 18, 269-285.
Copyright@2019 University of Chinese Academy of Sciences.All Rights Reserved.
京ICP备19028953号-1